Mapping long reads onto my references: the first time were darn headaches. Jargon, acronyms, concepts I barely grasped. Then, I created this section of my computational analysis of DNA sequencing datasets (Part II).
In this post, we’ll see, among the others:
- SAM, BAM, CRAM
- MAPQ scores and smoke-free CIGARs
- The 3 types of alignments.
I hope this short write-up spares you the headaches I had.
REMAINDER: This glossary is divided in three parts, covering the three major computational steps in the analysis of DNA sequencing data:
- Reading,
- Mapping,
- Assembling a genome.
2. MAPPING A GENOME
08 – MAPPING: the computational process of aligning the sequencing reads to the best matching location(s) on the reference sequence. This process should be able to tell you how closely your reference matches the genome you have sequenced. The results of mapping are stored in SAM and BAM files.
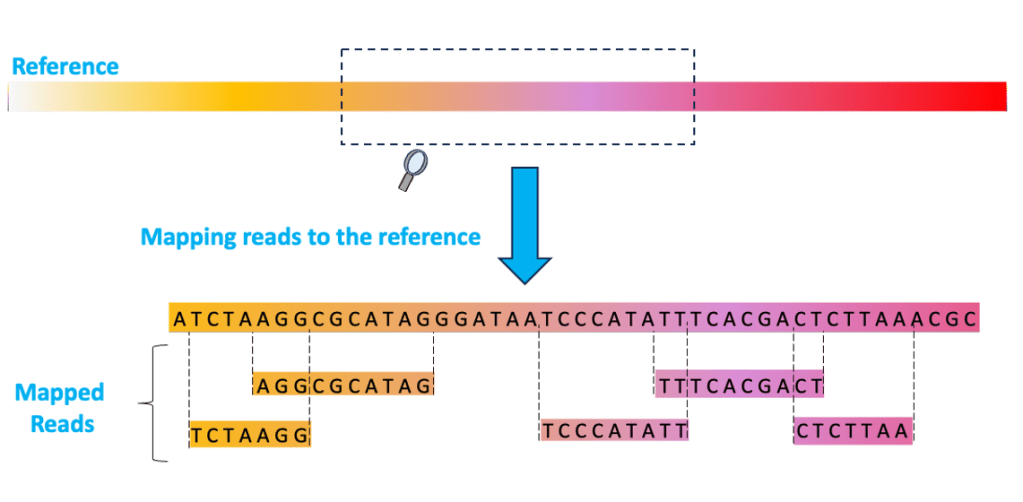
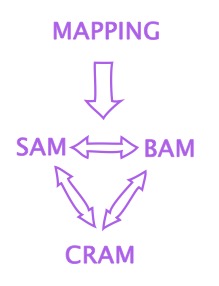
09 – SAM (Sequence Alignment Map): a text file that contains the results of the mapping process in a format that humans can theoretically read. For each mapped read, the SAM file lists the sequence of the read, where it maps (the genetic coordinates on the reference sequence), how (the CIGAR string), how well (the MAPQ score), if the read is primary, secondary, supplementary and more.

10 – BAM (Binary Alignment Map): a SAM file that was converted into a string of 1s and 0s (binary format) and then compressed. A BAM file is smaller and can be accessed faster by computers than the equivalent SAM file. In short: SAM is human-friendly, BAM is computer-friendly.
11 – CRAM (Compressed Reference-oriented Alignment Map): a highly-compressed SAM/BAM file that stores information on the mapping as a function of the reference sequence. Instead of listing every base in every read and how it maps, a CRAM file only retains information on which bases the read and the reference differ. As such, a CRAM file is useless if the reference is lost.
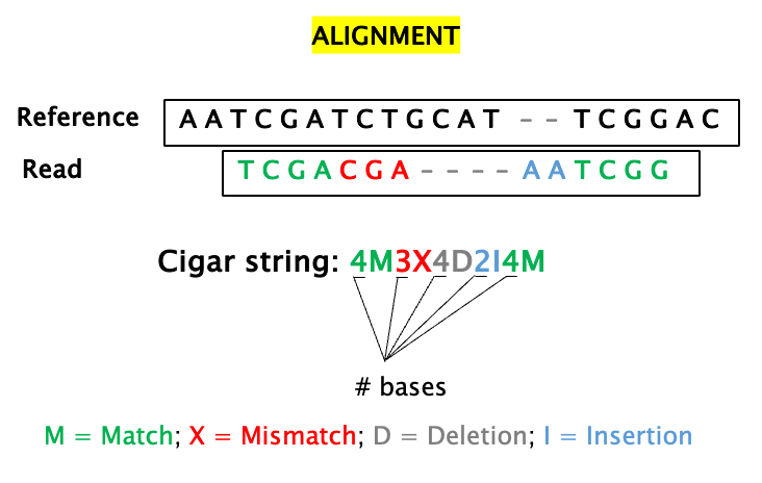
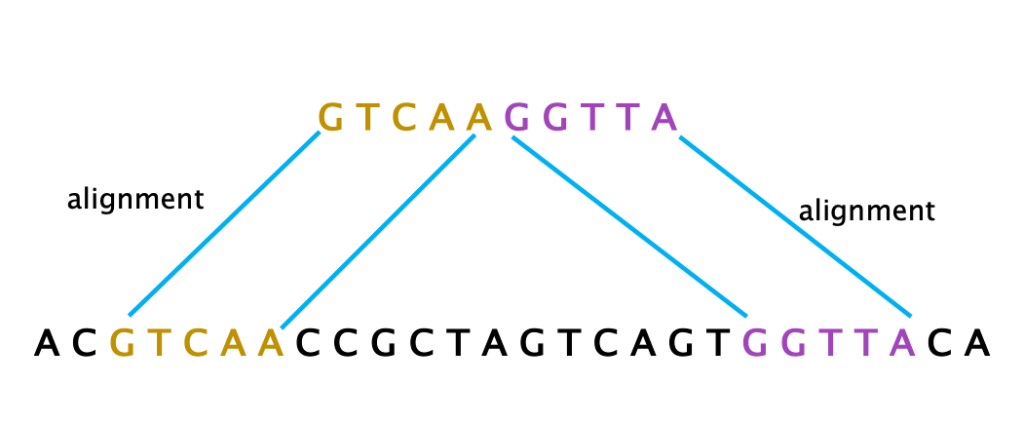
12 – CIGAR (Compact Idiosyncratic Gapped Alignment Report): a string of letters and numbers that summarises how a read aligns to a reference.
13 – MAPPING QUALITY (MAPQ) SCORE: integer metric that indicates the probability of a read mapping onto a specific location of the reference sequence. This is the confidence that the read derives from sequencing of that genetic place. Reads matching multiple locations (multimapping) have poor MAPQ score.
14 – PRIMARY ALIGNMENT: the single “best” matching alignment to the reference sequence (read align to that location with the highest MAPQ score).
15 – SECONDARY ALIGNMENT: an alternative alignment for a read mapping onto multiple locations (multi-mapping) of the reference. This often occurs when sequencing repetitive reads.
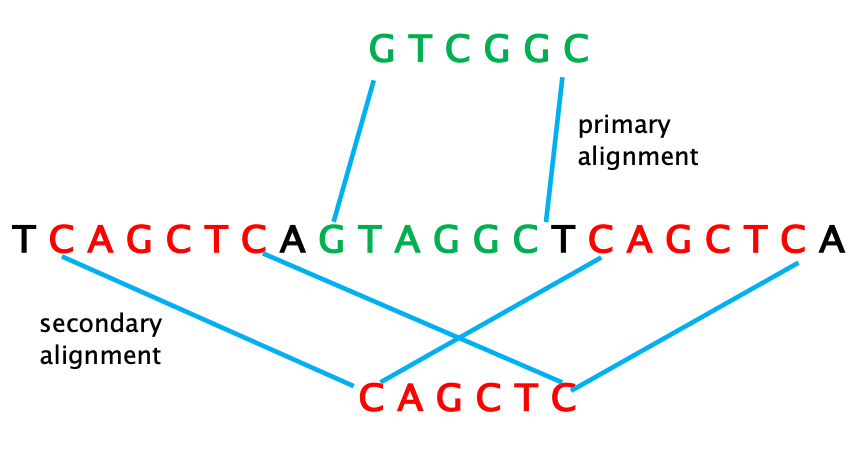
Note: even reads with secondary alignments have a primary alignment, which is the location of the reference sequence that matches them the best).
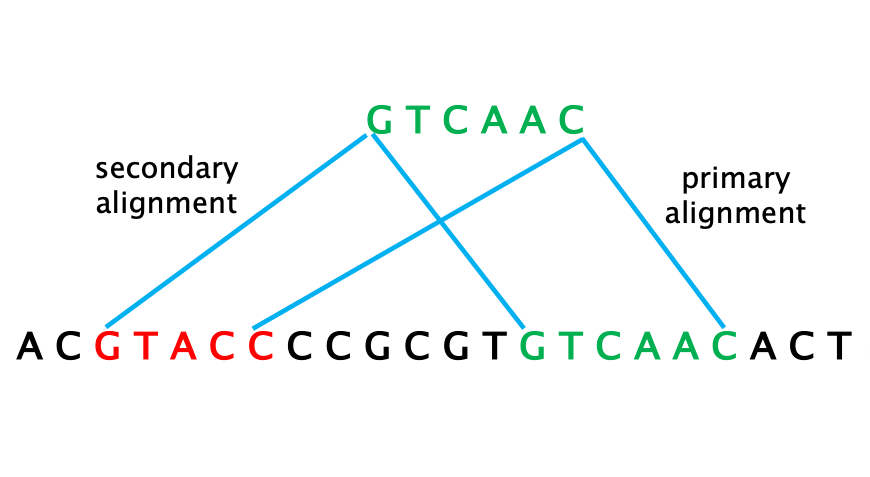
16 – SUPPLEMENTARY ALIGNMENT: When a single read is “split” in two (or more) parts that map onto two (or more) separated locations of the reference. This is indicative of a structural variation, such as an inversion, insertion or deletion, that has physically moved or joined parts of the genome.
WHAT’S NEXT
Time to stich reads together and assemble your genome.
- Will you use a reference-based approach or will you go de novo?
- How do sequencing coverage, depth and N50affect your confidence in the assembly?
- And what’s the difference between a contig and a scaffold?
We’ll discuss these points in PART III of this Computational Glossary of DNA Sequencing!