
I wrote reports about genome sequencing of genetically engineered viruses for years. And then, one day, I began doing the analysis myself (I was lucky enough to have a manager, Aaron Clack, believing in my abilities).
As I was immersed in the computations — with gratitude to Linta Kuncheria for laying the groundwork, and to Sylwia Jones & Praseed Grover for expertly doing the actual sequencing — I condensed the key ideas of the analysis into a glossary.
This brief write-up has helped me (still does!) reflect on my work and explain it. I hope you’ll find it useful too!
This glossary is divided in three parts, covering the three major computational steps in the analysis of DNA sequencing data:
- Reading,
- Mapping,
- Assembling a genome.
1. READING A GENOME
01 – BASECALLING: the computational process of converting the raw signals generated by sequencing a fragment of a sequencing library (the hundreds of thousands or even millions of snippets in which the target genome was split) into a sequencing read.

02 – SEQUENCING READ: the digital representation, produced by basecalling, of the nucleotides in a DNA fragment.
03 – FASTQ: a text file listing the reads that the sequencer has called. Every read is labelled with a unique identifier and all the nucleotides are accompanied by their own Phred quality scores. These information are arranged over four lines.
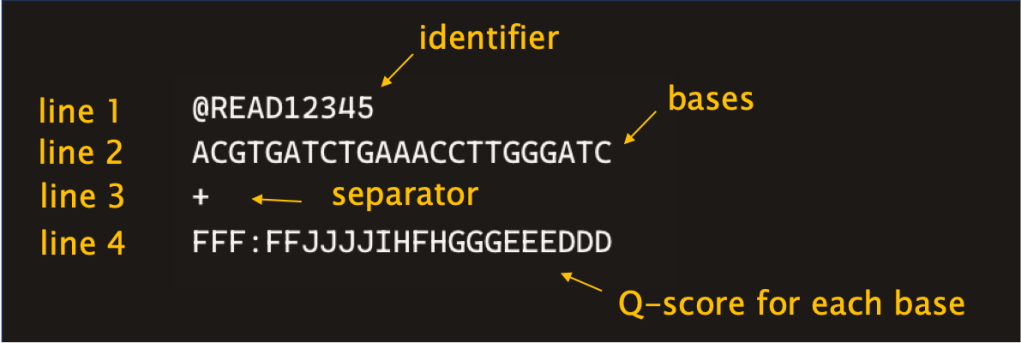
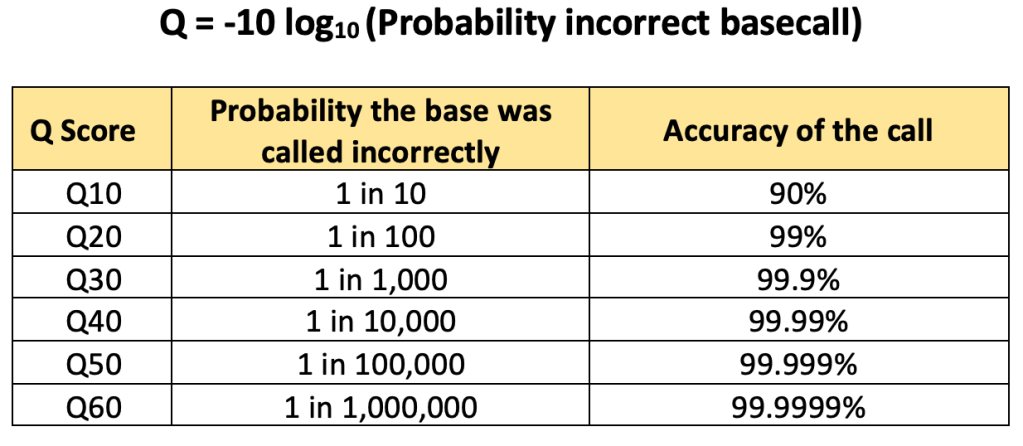
04 – Phred quality score (Q score): an integer metric indicating the probability that a base was called incorrectly. The higher the Q score, the more confident the sequencer is about the basecall. The average Q score of all the bases in a read is the Q score of that read. Reads with poor a Q score are filtered out of the FASTQ file to clean-up data before downstream analysis. The probability of incorrect basecall is higher in homopolymers (higher uncertainty in the actual sequence).
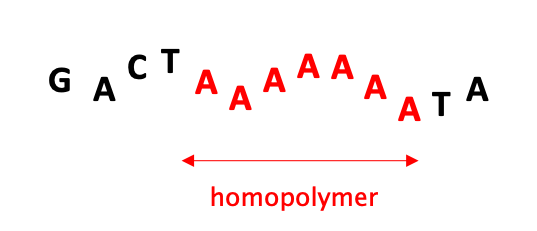
05 – HOMOPOLYMER: a continuos stretch of the same base repeating itself. Sequencers struggle with counting the number of bases in a homopolymer because the raw signal doesn’t change when reading these stretches. This uncertainty results in lower Q scores for those bases in a homopolymer.
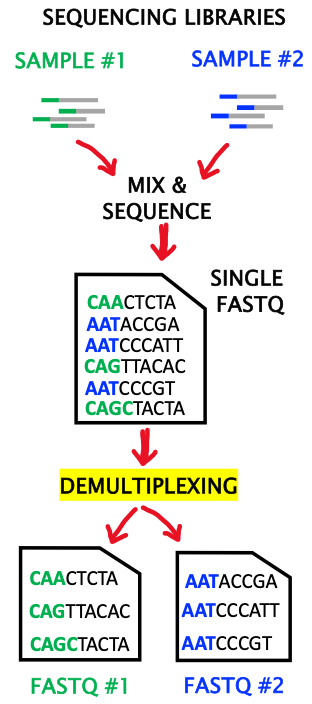
06. DEMULTIPLEXING: the computational process of sorting reads by sample of origin based on the barcode attached to the read (e.g.: reads generated by sequencing of treated vs untreated samples). Demultiplexing yields a new FASTQ file per barcode.
07 – REFERENCE SEQUENCE: a digital sequence believed to closely match the biological sequence of the genome being studied. It acts as a “map” to stitch together reads in the right order, but not all genomes have a reference.
WHAT’S NEXT
Once the sequencer has basecalled the DNA fragments into reads and compiled them into a FASTQ file—properly filtered by Q score and demultiplexed, the mapping to a reference sequence can begin (Part II). This process should be able to tell you how closely your reference matches the genome you have sequenced.
If a reference is not at hand, you may need to first assemble the genome from scratch (Part III).

Leave a comment